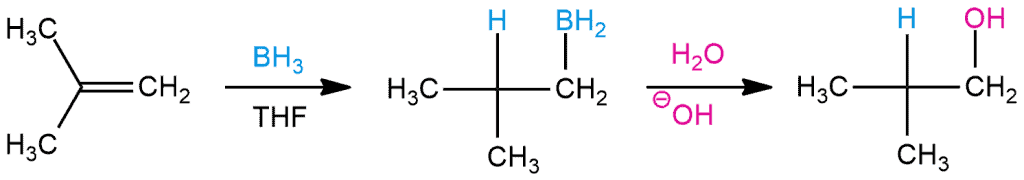
Hydroboration-oxidation is a reaction that converts an alkene to an alcohol. This reaction consists of two parts – hydroboration ( the addition of borane, BH3 to an alkene, forming an alkylborane) and oxidation (converts the C–B bond of the alkylborane to a C–O bond). Hydroboration-oxidation results in addition of H2O to an alkene.
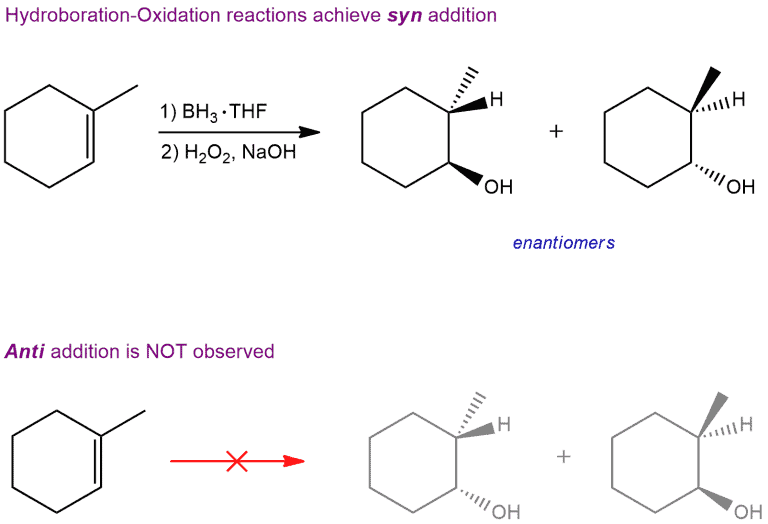
The stereochemical outcome of this reaction is also of particular interest. Addition of water (H and OH) leads to the formation of two chiral centers, and H and OH are added to the same side of the π bond. This mode of addition is called a syn addition. Only two of the four possible stereoisomers are formed. That is, the reaction doesn’t produce the two stereoisomers that would result from adding H and OH to opposite sides of the π bond (anti addition).
Another crucial thing is regioselectivity. This tells us which carbon will add to the hydroxide (actually boron), and which carbon will add hydrogen. Given that an anti-Markovnikov product is obtained, the hydroxide is added to the less substituted carbon.
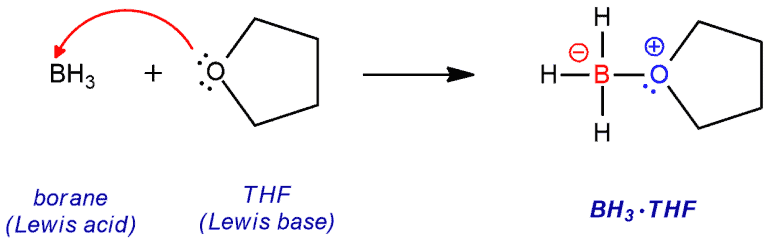
The structure of borane, BH3 is similar to that of a carbocation, but without the charge. The boron atom lacks an octet of electrons and is therefore very reactive. In fact, one borane molecule will even react with another borane molecule to form a dimeric structure called diborane. In these solutions, borane exists as a Lewis acid-base complex with the ether oxygen of THF (tetrahydrofuran), an aggregate that allows the boron to have an electron octet. THF can donate electron density into the empty p orbital of boron. Regardless, it is nevertheless still very electrophilic and subject to attack by the π bond of an alkene.
Mechanism of Hydroboration-oxidation
STEP 1. The borane adds to the double bond of an alkene. Because boron is sp² hybridized here, it’s a trigonal planar with empty p orbital which is capable of accepting a pair of electrons. The boron has about six electrons around it and the lack of octets makes it very reactive. In the presence of an alkene, boron gets closer to the carbon on the right side (less substituted) because other carbon has methyl group which is relatively bulky. The π electrons will attack the empty p orbital of the boron and form a bond between the carbon on the right side of the double bond and the boron. And as that bond is formed, it withdraws electron density from the carbon on the left and that carbon becomes partially positive (δ+). Increasing the partial positive charge on that carbon will trigger a hydride shift. A partial positive charge will attract hydrogen which will take the electrons of the B-H bond and form a bond between hydrogen and carbon on the left side.
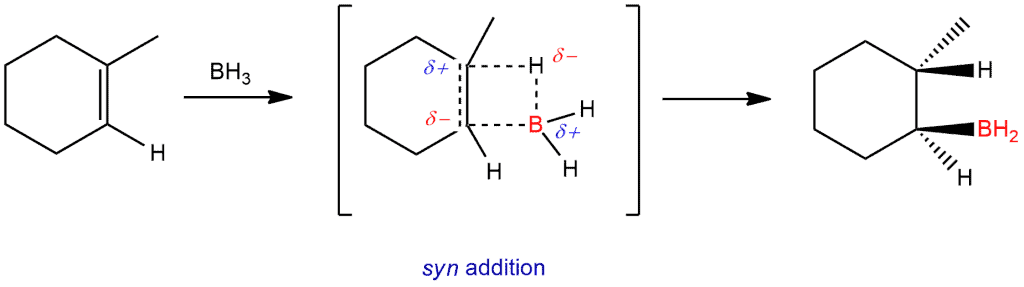
Depending on the literature, you can find that this step occurs once (as shown here) or three times to replace each hydrogen with an alkyl. Thus you can get trialkylborane.
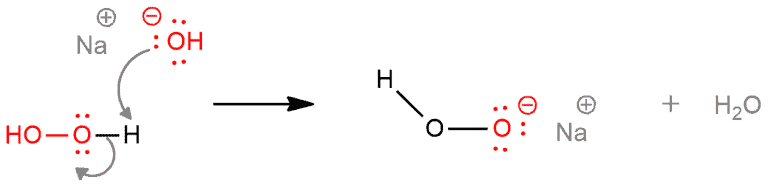
STEP 2. Deprotonation of hydrogen peroxide by the base. Hydroxide function as a base and it takes a proton from hydrogen peroxide, leaving electrons behind on the oxygen.
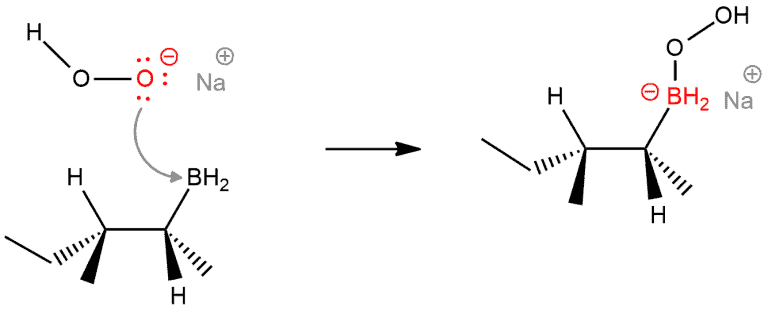
STEP 3. Nucleophilic attack of peroxide ion to alkylborane. This is a simple Lewis acid-base reaction. The deprotonated peroxide anion then adds to the empty orbital of boron, forming a negatively charged boron species. The boron-oxygen bond is formed.
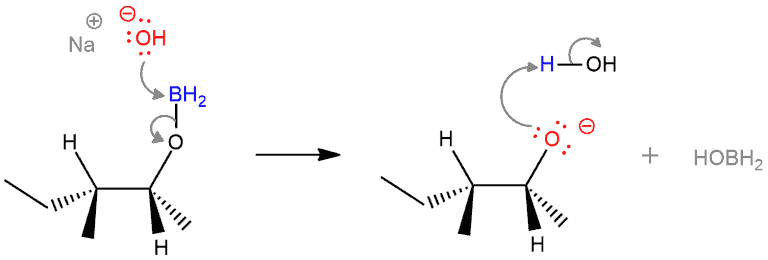
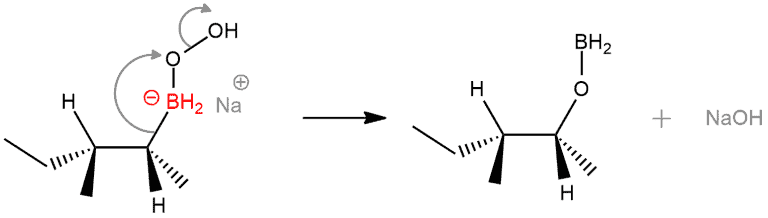
STEP 4. This step is a migration of an alkyl group. The pair of electrons in the C–B bond migrates to oxygen, leading to breakage of C–B and formation of C–O. The O-O bond is weak and also breaks down, forming hydroxide. This migration is very similar to 1,2-hydride and alkyl shifts we’ve seen previously, except that instead of migrating to the empty p orbital on a carbocation, the electron pair is essentially performing an attack on the σ* orbital of the O–O bond. The charge on boron goes from negative to neutral.
STEP 5. Nucleophilic attack of hydroxide anion and protonation of alkoxide. Hydroxide ion function as a nucleophile and attacks the empty p orbital of boron, and the O–B bond breaks. And the negatively charged oxygen is then protonated by water (the solvent).