Infrared (IR) spectroscopy is a very useful method for detecting the characteristic bonds of many functional groups through their absorption of infrared light.
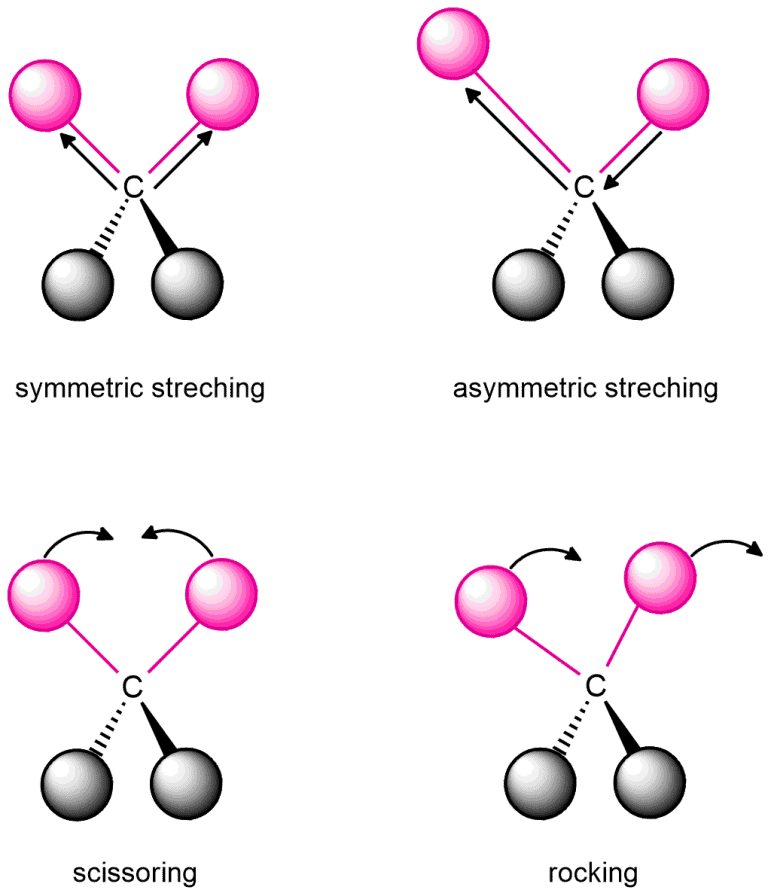
If you shine infrared light on a molecule, it is possible that the molecule absorbs energy from light. Absorbed energy can cause a bond to stretch or bend. We call that a stretching or bending vibration. These vibrations occur only at specific frequencies, which correspond to the frequency of IR light. When the frequency of IR light matches the frequency of a particular vibrational mode, the IR light is absorbed, and you can tell which frequencies are absorbed by looking at your infrared spectrum. Different kinds of bonds vibrate at different frequencies, so they absorb different frequencies of IR light, so it is possible to determine the functional groups present.
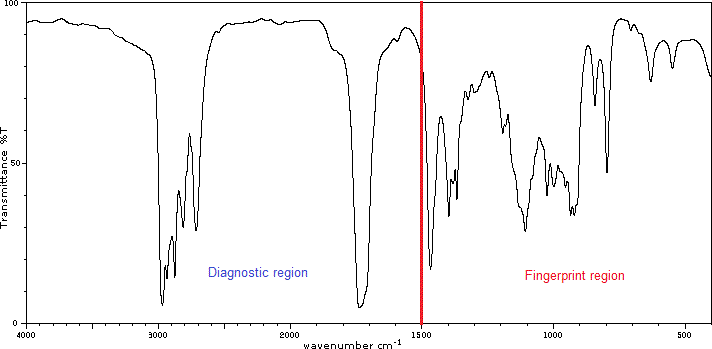
At first glance, the IR spectra look very complicated, but the only three things you need to know are:
- regions of the spectrum,
- one number (1500), and
- location and shape of the peaks.
Let’s considers all of these.
IR spectra can be divided into two main regions:
- Diagnostic region – generally has fewer peaks and provides the clearest information. This region contains all signals that arise from all bonds in a molecule.
- Fingerprint region – contains signals resulting from the vibrational excitation of most single bonds (stretching and bending).
Since the fingerprint region generally contains many signals and is more difficult to analyze, we can ignore it. It benefits us when we have similar compounds, for example, the same bonds (functional groups) but a different number of them. Such spectra will be the same in the diagnostic region, but in the fingerprint region won’t. Thus this region is called a fingerprint because each compound has a unique pattern of signals in this region, much the way each person has a unique fingerprint.
How will you distinguish these regions except the look of the peaks?
Using the value of the wavenumbers.
The abscissa of our IR diagrams shows the wavenumbers, and the boundary is at a value of 1500 cm‾¹. So we can also draw the line at a value of 1500 cm‾¹ when we interpret spectrum.
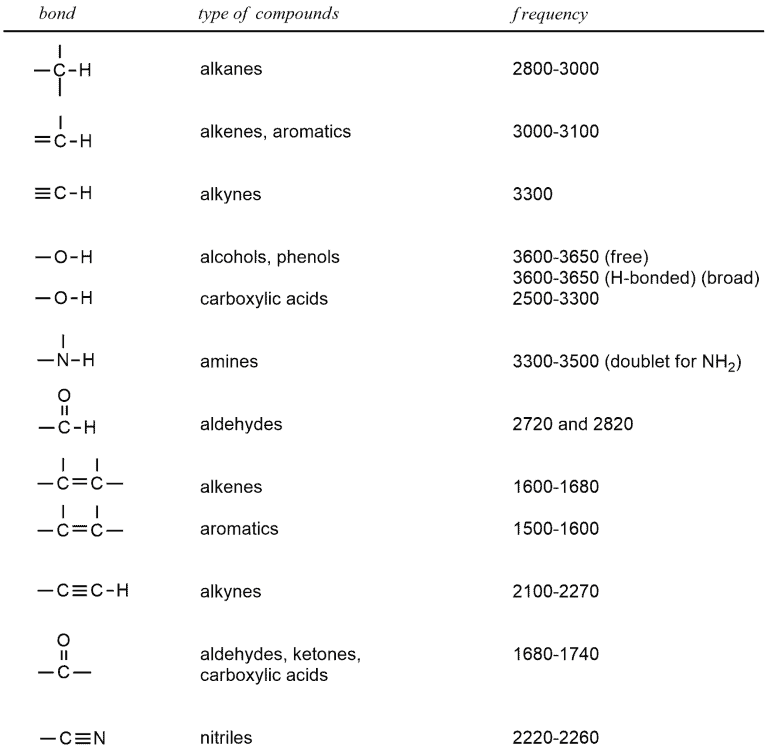
When we look at the following table of the characteristic stretching wavenumber values for the bonds, we can see that the most absorbing in the region above 1500 cm‾¹ and up to 3650 cm‾¹. This table shows some of the bonds and areas in which they appear. Some? So there are more? Of course, but this is enough to start. You’re probably confused with so many values in this table but don’t worry, you’ll easily remember them. You don’t have to learn all of these numbers right away because it will soon become a routine.
The third point explains everything. Now everything will be much much easier.
Now, all you need to learn is the location and shape of the peaks. Here are typical infrared absorption values for various types of bonds:
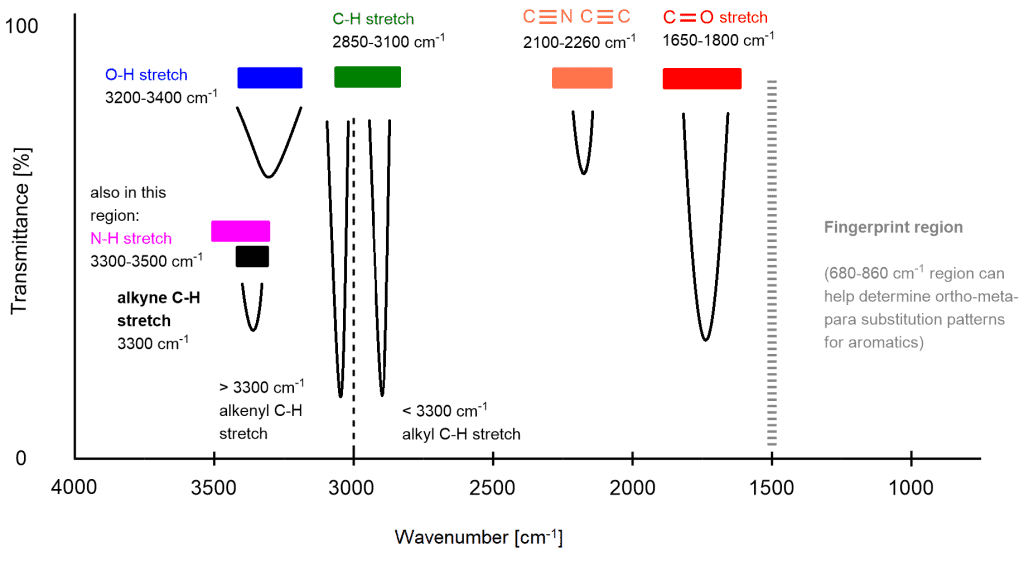
The most common signals are shown in the picture above, those you need to master first, but I will add even more (marked with *) in the text that also often appears in the spectra.
- If start at 1500 cm‾¹, the first thing we encounter is a very sharp needle a signal that is a carbon-carbon double bond.*
- In the region around 1700 cm‾¹, we can see a little bit thicker finger-like carbon-oxygen double bond.
- A little bit further, we might have the aromatic overtones that look like fangs. Their size is not clearly defined. They can be both short and long.*
- Then, we can see a very sharp needle-like signal that could correspond to triple bonds both carbon-carbon and carbon-nitrogen.
- Now, we have a signal that is really hard to interpret, and this is an aldehyde. Sometimes this signal occurs but actually, we don’t have an aldehyde. Because it is necessary to check ¹H NMR if this compound is present or not.
- A little before 3000 cm‾¹ we expect sp³ hybridized carbon atom attached to hydrogen. Sometimes they are smaller or larger than other signals.
- And a little bit after 3000 cm‾¹ we expect sp² hybridized carbon atom attached to hydrogen.
- After sp³ and sp² comes the sp hybridized carbon atom bonded to hydrogen. If we interpret that we have some of these hybridized carbon atoms, we must confirm them with the previously mentioned signals. In other words, if we say that we have signal number 8. we also have signal number 4. And if we have signal number 7. we also have signal number 1.
- Then, further past 3000 cm‾¹, we encounter with the large signal of a hydroxyl group. The OH stretch shows up in this region is a large singlet because it’s more acidic.
- In the same region as the OH, we might have an NH stretch. The NH may show a singlet, a doublet or even a triplet sometimes which depends on whether we have a primary or secondary amine. These signals are more shorter than OH signal.
- In the region where the triple bond exists, we can have a shorter version of that which is the NH Bend. This occurs for primary amines and amides.
- And for the end, carboxylic acids. This signal appears from OH stretch to between an aldehyde and a triple bond. This signal is very wide and short.
I’m a former science teacher, and I was actually looking for where carbon dioxide absorbs. I don’t think CO2 is considered a ketone, right? Do the C=O bonds in CO2 absorb near 1700? or 1650? or 1800? I’d also like to know what happens to the absorbed energy next? Do I need an IR emission chart to find out?
Thank you for your comment. Here’s an IR chart for CO2: https://webbook.nist.gov/cgi/cbook.cgi?ID=C124389&Type=IR-SPEC&Index=1
Because of the two C=O bonds, the shift is ~2300 cm -1.